Usage options¶
mrbait reads all options and inputs using command-line arguments provided after the program name. For a quick look at all options from the command line, call the help menu by typing mrbait -h from the terminal.
Note that options requiring a floating point number (e.g. -q) allow inputs from 0.0 to 1.0, and options requiring an integer (e.g. -c) allow inputs ranging from 1 to infinity.
Main Parameters¶
General options¶
| -r, --resume | Resume: This flag is used to tell mrbait if you would like to resume work Usage: |
| --db | Database: Use this with the –resume flag to specify a .sqlite database file from which to start the pipeline. |
| -T, --threads | Threads: Number of threads to use with processes that run in parallel. This will also be passed to vsearch and/or blast if those are being called. [default=1] |
| -h, --help | Help: Exit and display the help menu |
Input Options¶
| -M, --maf | MAF input: Use this to provide the path to the multiple alignment MAF file |
| -X, --xmfa | XMFA input: As an alternative to the MAF file, you can provide the .xmfa file output by the aligner Mauve. |
| -L, --loci | LOCI input: Multiple alignments can also be provided using the .loci file output by the RADseq assembly pipeline pyRAD. |
| -A, --assembly | FASTA input: Genome assembly provided as FASTA |
| -V, --vcf | VCF input: For use with –assembly: VCF file containing variant data |
| -G, --gff | GFF input: For use with –assembly: GFF file containing feature data |
| --vcfALT | REF calling with VCF: For use with –assembly and –vcf: This option tells mrbait to attempt to override N/gap characters in the reference sequence using values from the ALT column in the VCF file. [default=off; boolean] |
Alignment filtering/ consensus options (use with -M, -X, -L)¶
| -c, --cov | Coverage: Minimum number of individuals/sequences per alignment, for MAF, XMFA, or LOCI inputs [default=1] |
| -l, --len | Minimum length: Minimum alignment length to attempt bait design [default=80] |
| -q, --tresh | Bad base threshold: Threshold proportion of gaps or N (ambiguous or poor quality) characters to over-ride the consensus base. For example, -q 0.2 would be interpreted as 20% of bases at a nucleotide position must be an “N” or gap character in order for that character to be represented as the consensus base. [default=0.1] |
| -Q, --max_ambig | |
| Max bad bases: Maximum allowable proportion of gap/N characters allowed in a consensus sequence before it will be discarded. -Q 0.5 means a consensus sequence can be 50% N’s or gap characters (“-“) before being dropped from consideration. [default=0.5] | |
| -k, --mask | Mask threshold: Threshold proportion of masked characters per nucleotide column to mask the consensus base call. For use when case represents masking information (where lowercase = masked), as when using the -xsmall option in RepeatMasker to flag low-complexity or repetitive sequences. Case will be retained in the consensus on a per-base basis according to this threshold. [default=0.1] |
| -K, --max_mask | Max masked bases: Maximum allowable proportion of masked characters If lowercase bases do not contain masking information, set to -K 1.0 |
| --dustMask | Perform DUST masking: Using this flag will have mrbait mask the consensus sequences using the DUST algorithm in VSEARCH |
General Bait Design Options¶
| -b, --bait | Bait length: This is the length of desired baits, and will be used for bait design as well as the sliding window width for target region discovery [default=80] |
| -w, --win_shift | |
| Sliding window shift distance: Shift distance for sliding window used to discover target regions. Generally, there should not be a reason to alter this. If target discovery (step 2) is taking a very long time, adjusting this may make it faster although it could result in more targets failing filtering [default=1] | |
| -v, --var_max | Maximum SNPs per bait: Maximum allowable variants allowed in a bait sequence. These can be expanded in the final output as each possible non-ambiguous bait sequence for synthesis. Use this when there are not enough conserved regions to capture enough loci for your design. [default=0] |
| -n, --numN | Maximum Ns per bait: Maximum allowable ambiguous (N) bases allowed per bait. This could be increased when there are too many poor quality bases in your alignment to design a sufficient number of probes, although keep in mind this will affect the specificity of your resulting probes. [default=0] |
| -g, --numG | Maximum gaps per bait: Maximum allowable gap characters allowed per bait. If dealing with alignments containing many indels, it might be desirable to allow a small number per bait sequence. These can be expanded in the final output using the -x,–expand option, which will expand gap characters as A, G, T, C, and absent. [default=0] |
Target Region Options¶
These are options primarily used to control which target regions, or regions which could be enriched (e.g. conserved enough to design baits) will be used to design the final set of bait sequences. Targets will be either passed or failed depending on these criteria.
For example, you can constrain targets to fail if they are below 200 bases in length, or above 5000 based in length by specifying -F len=200,5000. In this case, all targets with total lengths outside of this range will fail, and be excluded from bait design.
| -R, --mult_reg | Multiple targets per locus: By default, mrbait only chooses one target region (e.g. conserved region for which baits could be designed) per locus/ alignment. When multiple are discovered, they are ranked according to the criterion selected with the -S,–select_r option. When -R,–mult_reg not in use, only a single target region (and corresponding baits) is chosen per alignment. [default=false] |
| -m, --min_mult | Minimum length for multiple targets: Specify this to set a minimum alignment or locus length to allow multiple target regions to be selected. By default will be set to the value of -l,–len (thus, when -R,–mult_reg is used, all loci passing length filter will be allowed multiple targets). |
| -D, --dist_r | Distance between targets: When -R,–mult_reg is in use, use this parameter to specify the minimum distance between targets. When targets are in conflict (e.g. they are less than -D,–dist_r bases apart), conflicts will be resolved using the criterion set with -S,–select_r. [default=100] |
| --target_all | Use all loci as targets: This option tells mrbait to use all passing loci as target regions for bait design. Note that all target filtering options will still be in effect. Use this option if your input loci are already curated, and you simply want to design baits for them [default=False] |
| -d, --flank_dist | |
| Flanking distance for target filtering: Distance from boundaries of target region to parse for counting SNPs, ambiguities, gaps, etc when filtering target regions (see -S,–select_r and -F,–filter_r) [default=500]. Note that this value will tell mrbait to search ‘d’ bases to the left AND right of each target region. Note that currently, the same –flank_dist value will be used for all filters. | |
| -S, --select_r | Target selection criterion: Method to resolve conflicts when targets are Usage:
-S snp: Select target with most SNPs within d bases Example: -d 100 -S snp to choose region with most SNPs within |
| -F, --filter_r | Target filtering criteria: Method(s) used to filter all target regions. Usage:
-F gff=[type]: Only retain targets within d bases of a GFF-annotated feature of type type. Only for use when -A and -G inputs provided. Use -F gff=all to target any type of annotated feature. Examples: |
Bait Selection Options¶
These options are used to specify how baits will be designed in passing target regions (-s, –select_b) and how designed baits will be curated to create the final set of sequences for synthesis (-f, –filter_b).
The default behavior is to tile baits across all passing target regions, with an overlap of 50%. This corresponds to ~2X coverage across the target region. Only passing baits will be included in the final output FASTA file.
| -s, --select_b | Bait selection scheme: Use this to specify the desired method Usage: |
| -f, --filter_b | Bait filtering criteria: Method(s) used to filter baits. Usage: |
Output Options¶
Use these options to control the format of your output file, or to specify non-default output files.
| -x, --expand | Bait format: Boolean. Use this flag if you want any ambiguities in bait sequences to be expanded (e.g. N = A,G,C,T). IUPAC codes will be fully expanded, and gap characters will be expanded as all nucleotides and as absent. Bait sequences will be output as FASTA to $out_baits.fasta |
| -t, --print_tr | Print target regions: Boolean. Use this flag if you would like target regions to be printed to a FASTA file. FASTA headers will reflect locus number, target number within locus, and pass=T or pass=F indicating if the target passed or failed filtering specified using the -F,–filter_r options, as|br| well as any targets which were excluded due to -S,–select_r criteria. Output file will be named as $out_targets.fasta. |
| --print_loc | Print locus cataog: Boolean. Prints consensus loci, formatted as in –print_tr |
| --strand | Output strand: Use this if you want to print baits as-designed, or as reverse complement. Possible values: “+” [default], “-” (reverse-complement), or “both”. |
| -o, --out | Output prefix: Desired prefix for output files. Default is “out”. |
Filtering using vsearch¶
Using the –filter_r or –filter_b ‘pw’ (for pairwise-align) options call an external open-source package vsearch. Internally, this is accomplished using the “allpairs_global” function in vsearch, which performs all-vs-all global alignments of target or bait sequences. mrbait then parses the output of vsearch to find pairs of sequences with greater than i percent identity over at least q percent of the query sequence. Sequences are first sorted by length, meaning the q proportion should be measured by the shorter of the two sequences in each pairwise alignment (although when called with the –filter_b command, all pairs are of equal length). Because all sequences will be compared in a pairwise fashion, keep in mind that this step could take considerable time, although VSEARCH is exceptionally efficient and does take advantage of multiple cores for parallel computation (passed using the –threads argument).
If you did NOT install vsearch using the conda installation instructions for mrbait, you may need to point mrbait to the vsearch executable. You can specify this path, as well asspecify how mrbait parses the results of vsearch using the following parameters:
vsearch Options¶
| --vsearch | VSEARCH binary: Path to vsearch binary. If installed via conda install, the bioconda recipe will be used, and placed into the bin/ folder for your conda installation. mrbait, by default, assumes that the vsearch executable is locatable in your $PATH as “vsearch”. If this is not the case, provide an alternate path using this flag. |
| --vthreads | VSEARCH threads: Number of threads to use for vsearch. By default, this is assumed to be the same value as the –threads argument passed to mrbait |
| --noGraph | No conflict graph: By default, mrbait will try to recover as many sequences as possible from the pairwise alignment results by using a graph structure (see below) to remove only enough sequences to resolve all conflicts |
| --noWeightGraph | |
| Unweighted conflict resolution: By default, mrbait weights all nodes by the number of SNPs that the corresponding target or bait captures. Use this option to turn this off, and instead use the built-in maximal_independent_set function from the networkx package. | |
| --weightByMin | Weight by minimum ambiguity: Instead of trying to keep sequences with the most flanking SNPs, this option tries to keep sequences with the fewest gap or N characters in flanking sequences. |
| --weightMax | Maximum size to attempt weighting: Maximum graph size to attempt weighted algorithm for finding maximal independent set. Graphs larger than this size will use the maximal_independent_set function from the networkx package, which is slightly faster. |
Graph-based conflict resolution¶
After vsearch is complete, mrbait will parse the output and represent all ‘conflicting’ sequences (e.g. those which aligned with i or greater identity and q or greater query coverage) as a graph structure, with nodes as the primary key (ID) for the SQLite entry corresponding to each sequence, and edges unweighted and representing ‘conflicts’ from the pairwise alignment. By default, mrbait uses this graph structure to attempt to rescue as many sequences as possible which failed the pairwise alignment filter by using an algorithm to find the most sequences which can be “kept” while removing all edges (conflicts) from the graph (e.g. see Figure 2 below).
This is accomplished by searching through all edges, removing the node (target or bait) which captures the least flanking SNPs in the original alignments, or if this value is equal by removing the node with the most neighbors (=conflicting sequences). If both quantities are equal, one is chosen at random. Options are provided to turn off this behavior altogether, or to weight nodes according to how many gap/N characters flank them, or to alternatively use the maximal_independent_set function from the networkx package. Unfortunately, finding the maximal independent set of a graph (i.e. the maximum number of nodes to retain while removing all edges) is an extremely hard problem, and cannot be done in a particularly efficient manner. Thus, with very large sets of target sequences, this can take quite a while if there is a lot of highly matching alignments among them.
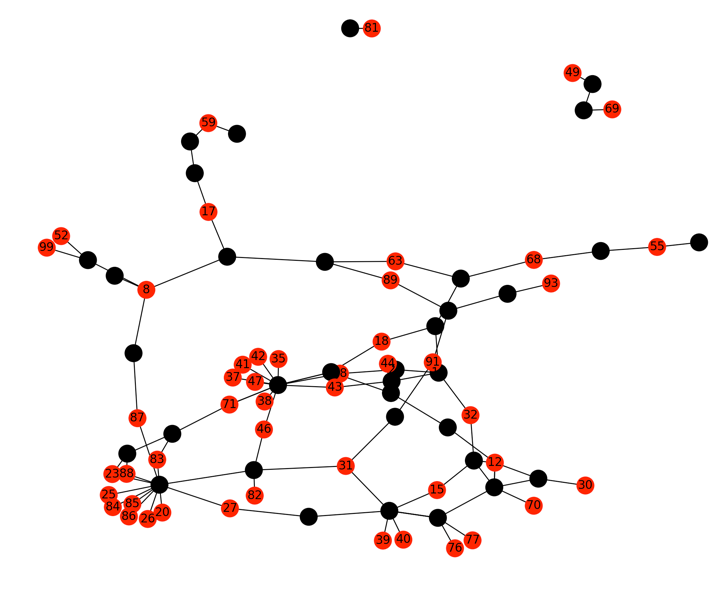
Figure 2: Example maximal independent set (plotted using the matplotlib for Python!) where nodes in red are those which have been kept by the algorithm. Black nodes represent sequences which would be deleted (e.g. baits which fail the ‘pw’ filter). Although red nodes may be indirectly connected by black ‘failed’ nodes (i.e. by multiple edges), no direct edges remain in the graph after removing failed nodes.
Filtering using blast¶
The NCBI-BLAST+ package (https://blast.ncbi.nlm.nih.gov/Blast.cgi) can also be called using a similar scheme of setting a threshold of i sequence identity of hits, and q query coverage. Currently, mrbait supports blastn filtering using these cutoff values to either exclude (blast_x) or include (blast_i) sequences based on presence of hits.
Usage of the ‘blast_x’ option for either target (–filter_r) or bait (–filter_b) filtering would most generally be used to prevent non-target matches to potential contaminant DNA, given a representative genome for the putative non-target organism (for example a common bacterial contaminant). If such a reference genome is lacking, you can also increase specificity of chosen targets or baits using the ‘blast_i’ option, which only retains queries with hits to a specified genome.
When using either ‘blast_x’ or ‘blast_i’ options for filtering, a database file must be specified either as a pre-built BLAST-formatted database using the –blastdb flag, or alternatively as a FASTA file using –fastadb. In the latter case, mrbait will call the NCBI MakeBlastDB executable. When using the conda install, this is included with all other BLAST+ binaries (including the blastn executable also required by mrbait for BLAST filtering). If not using the conda installation instructions, these are both assumed to be accessible in your $PATH as ‘blastn’ and ‘makeblastdb’.
Please note that with particularly large sets of query sequences, this process may take a while, although it does take advantage of multithreading if passed via the –threads argument.
BLAST options:
| --blastn | BLASTN binary: Path to blastn binary. If installed via conda install, the bioconda recipe will be used, and placed into the bin/ folder for your conda installation. mrbait, by default, assumes that the blastn executable is locatable in your $PATH as “blastn”. If this is not the case, provide an alternate path using this flag. |
| --makedb | MakeBlastDB binary: Path to makeblastdb binary. If installed via conda install, the bioconda recipe will be used, and placed into the bin/ folder for your conda installation. Mrbait, by default, assumes that the makeblastdb executable is locatable in your $PATH as “makeblastdb”. If this is not the case, provide an alternate path using this flag. |
| --blastdb | Path to BLAST database: Full path to formatted database to search against |
| --fastadb | Path to FASTA database: If database is provided as a FASTA file, and requires building using the makeblastdb command, provide that path here. |
| --e_value | E-value threshold: Minimum e-value cutoff to report BLAST hits [default=0.000001] |
| --gapopen | Gap opening penalty: Penalty for opening gaps when performing alignment [default=5] |
| --gapextend | Gap extension penalty: Penalty for extending gaps when performing alignment [default=2] |
| --word_size | Word size: Word size [default=11] |
| --max_hits | Max hits: Value for BLAST parameter –max_target_seqs [default=10000] |
| --nodust | Turn off dusting: Use this flag to turn off the low-complexity filter using by blastn. Depending on what your goals are for BLAST filtering, this may be necessary. Boolean. |
| --megablast | Use megablast: Use this flag to switch to the megablast algorithm. It is recommended you use this if you are trying to exclusively find nearly identical alignments to a very closely related genome. |